
起こりうる副作用とその対策
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行ってください。
(1)重大な副作用
1)敗血症性ショック、敗血症(頻度不明※1)、肺炎(0.9%※1)等の重篤な感染症
- 本剤投与により、敗血症、肺炎、真菌感染症を含む日和見感染症等の致死的な感染症があらわれることがあります。
- 本剤は、細胞性免疫反応を調節するTNFαの生理活性を抑制するので、感染症に対する宿主免疫能に影響を及ぼす可能性があります。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 重篤な感染症(敗血症等)の患者さんに対しては、本剤は投与禁忌です。
- 感染症の患者さん又は感染症が疑われる患者さんに対しては、慎重に投与してください。
- 本剤とアバタセプト(遺伝子組換え)の併用は行わないでください。海外で実施したプラセボを対照とした臨床試験において、抗TNF製剤とアバタセプト(遺伝子組換え)の併用療法を受けた患者さんでは併用による効果の増強は示されず、感染症及び重篤な感染症の発現率が抗TNF製剤のみによる治療を受けた患者さんでの発現率と比べて高かったと報告されています。また、本剤と他の生物製剤の併用について安全性及び有効性は確立していないので併用を避けてください。
- 本剤による治療中は十分な観察を行い、感染症の発現や増悪に注意してください。
対処法
- 異常が認められた場合には本剤の投与を中止するなどの適切な処置を行い、感染症が消失するまでは本剤を投与しないでください。感染症により死亡に至った症例が報告されています。
- 重篤な感染症の初期症状、感染症が疑われる所見が認められる場合には、本剤による治療を一時中断し、「関節リウマチ(RA)に対するTNF阻害薬使用の手引き」を参考に適切な処置を行ってください。
1. 警告(抜粋)
1.1
本剤投与により、結核、肺炎、敗血症を含む重篤な感染症及び脱髄疾患の新たな発現若しくは悪化等が報告されており、本剤との関連性は明らかではないが、悪性腫瘍の発現も報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。また、本剤の投与において、重篤な副作用により、致命的な経過をたどることがあるので、緊急時の対応が十分可能な医療施設において医師の管理指導のもとで使用し、本剤投与後に副作用が発現した場合には、主治医に連絡するよう患者に注意を与えること。[ 8.1-8.3、9.1.1-9.1.3、11.1.1、11.1.3、11.1.4参照]
1.2 感染症
1.2.1
重篤な感染症 敗血症、肺炎、真菌感染症を含む日和見感染症等の致死的な感染症が報告されているため、十分な観察を行うなど感染症の発症に注意すること。[8.1、9.1.1、11.1.1参照]
2. 禁忌(次の患者には投与しないこと)(抜粋)
2.1
重篤な感染症(敗血症等)の患者[症状を悪化させるおそれがある。][8.1、11.1.1参照]
9.特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.1
感染症(重篤な感染症を除く)の患者又は感染症が疑われる患者 適切な処置と十分な観察が必要である。[1.1、1.2.1、8.1、11.1.1参照]
9.1.6
B型肝炎ウイルスキャリアの患者又は既往感染者(HBs抗原陰性、かつHBc抗体又はHBs抗体陽性) 肝機能検査値や肝炎ウイルスマーカーのモニタリングを行うなど、B型肝炎ウイルスの再活性化の徴候や症状の発現に注意すること。また、B型肝炎に関して専門知識を持つ医師に相談することが望ましい。本剤を含む抗TNF製剤を投与されたB型肝炎ウイルスキャリアの患者又は既往感染者において、B型肝炎ウイルスの再活性化が報告されている。報告された症例の多くは、免疫抑制作用をもつ薬剤を併用していた症例である。[8.4、11.1.1参照]
11. 副作用(抜粋)
11.1
重大な副作用
11.1.1
敗血症性ショック、敗血症(頻度不明)肺炎(0.9%)等の重篤な感染症 重篤な感染症及び真菌感染症等の日和見感染症があらわれることがある。また、B型肝炎ウイルスの再活性化があらわれることがある。異常が認められた場合には、感染症が消失するまで本剤を投与しないこと。なお、感染症により死亡に至った症例が報告されている。[1.1、1.2.1、2.1、8.1、8.4、9.1.1、9.1.6参照]
2)間質性肺炎(0.5%※1)
- 本剤投与により、間質性肺炎があらわれることがあります。
- 先行バイオ医薬品を含む抗TNF製剤の投与により、結核の発現が報告されており、中には致死的な例も報告されています。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 本剤を投与中は定期的に問診を行うなど患者さんを観察してください。
- 発熱、咳嗽、呼吸困難等の呼吸器症状の発現に注意してください。
- 間質性肺炎の既往歴のある患者さんでは、間質性肺炎が増悪又は再発する場合があるため、十分に注意してください。
対処法
- 異常が認められた場合には、速やかに胸部X線検査、胸部CT検査及び血液ガス検査等を実施し、本剤の投与を中止するとともにニューモシスティス肺炎との鑑別診断(β-Dグルカンの測定等)を考慮に入れ適切な処置を行ってください。
- 間質性肺炎が疑われる場合には、「関節リウマチ(RA)に対するTNF阻害薬使用の手引き」を参考に適切な処置を行ってください。
9.特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.5
間質性肺炎の既往歴のある患者 定期的に問診を行うなど、注意すること。間質性肺炎が増悪又は再発することがある。[11.1.2参照]
11. 副作用(抜粋)
11.1
重大な副作用
11.1.2
間質性肺炎(0.5%)発熱、咳嗽、呼吸困難等の呼吸器症状に十分注意し、異常が認められた場合には、速やかに胸部X線検査、胸部CT検査及び血液ガス検査等を実施し、本剤の投与を中止するとともにニューモシスティス肺炎との鑑別診断(β-Dグルカンの測定等)を考慮に入れ適切な処置を行うこと。[9.1.5参照]
3)結核(頻度不明※1)
- 本剤投与により、播種性結核(粟粒結核)及び肺外結核(胸膜、リンパ節等)を含む結核が発現し、致命的な症例も報告されています。
- 先行バイオ医薬品を含む抗TNF製剤の投与により、結核の発現が報告されており、中には致死的な例も報告されています。
- ツベルクリン反応等の検査が陰性の患者において、投与後活動性結核が認められた例も報告されています。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 活動性結核の患者さんに対しては、本剤は投与禁忌です。
- 本剤投与に先立って結核に関する十分な問診及び胸部X線検査に加え、インターフェロン-γ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認してください。必要に応じて「生物学的製剤投与時の結核予防対策」(次ページ)を参照してください。
- 本剤投与前にツベルクリン反応等の検査が陰性の患者さんにおいても、投与後活動性結核があらわれる場合があるため注意してください。
- 結核の既往歴を有する患者さん及び結核の感染が疑われる患者さんには、結核等の感染症について診療経験を有する医師と連携の下、原則として本剤の投与開始前に適切な抗結核薬を投与してください。
- 本剤投与中は、胸部X線検査等の適切な検査を定期的に行うなど結核の発現には十分に注意してください。
対処法
- 本剤投与後は十分に観察を行い、異常が認められた場合には本剤の投与を中止するなどの適切な処置を行ってください。
- 結核を疑う症状が発現した場合(持続する咳、体重減少、発熱等)には速やかに担当医に連絡するよう患者さんに指導してください。
1. 警告(抜粋)
1.1
本剤投与により、結核、肺炎、敗血症を含む重篤な感染症及び脱髄疾患の新たな発現若しくは悪化等が報告されており、本剤との関連性は明らかではないが、悪性腫瘍の発現も報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。また、本剤の投与において、重篤な副作用により、致命的な経過をたどることがあるので、緊急時の対応が十分可能な医療施設において医師の管理指導のもとで使用し、本剤投与後に副作用が発現した場合には、主治医に連絡するよう患者に注意を与えること。[8.1-8.3、9.1.1-9.1.3、11.1.1、11.1.3、11.1.4参照]
1.2 感染症
1.2.2
結核 播種性結核(粟粒結核)及び肺外結核(胸膜、リンパ節等)を含む結核が発症し、致命的な例も報告されている。本剤投与に先立って結核に関する十分な問診及び胸部X線検査に加え、インターフェロン-γ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。結核の既往歴を有する患者及び結核の感染が疑われる患者には、結核等の感染症について診療経験を有する医師と連携の下、原則として本剤の投与開始前に適切な抗結核薬を投与すること。ツベルクリン反応等の検査が陰性の患者において、投与後活動性結核が認められた例も報告されている。[8.3、9.1.2、11.1.3参照]
2. 禁忌(次の患者には投与しないこと)(抜粋)
2.2
活動性結核の患者[症状を悪化させるおそれがある。][8.3、11.1.3参照]
9.特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.2
結核の既往歴を有する患者又は結核感染が疑われる患者
(1)結核の既往歴を有する患者では、結核を活動化させるおそれがある。[1.1、1.2.2、8.3、11.1.3参照]
(2)結核の既往歴を有する場合及び結核感染が疑われる場合には、結核の診療経験がある医師に相談すること。下記のいずれかの患者には、原則として本剤の投与開始前に適切な抗結核薬を投与した上で、本剤を投与すること。[1.1、1.2.2、8.3、11.1.3参照]
- 胸部画像検査で陳旧性結核に合致するか推定される陰影を有する患者
- 結核の治療歴(肺外結核を含む)を有する患者
- インターフェロン-γ遊離試験やツベルクリン反応検査等の検査により、既感染が強く疑われる患者
- 結核患者との濃厚接触歴を有する患者
11. 副作用(抜粋)
11.1
重大な副作用
11.1.3
結核(頻度不明) 結核(播種性結核、肺外結核を含む)があらわれることがある。[1.1、1.2.2、2.2、8.3、9.1.2参照]
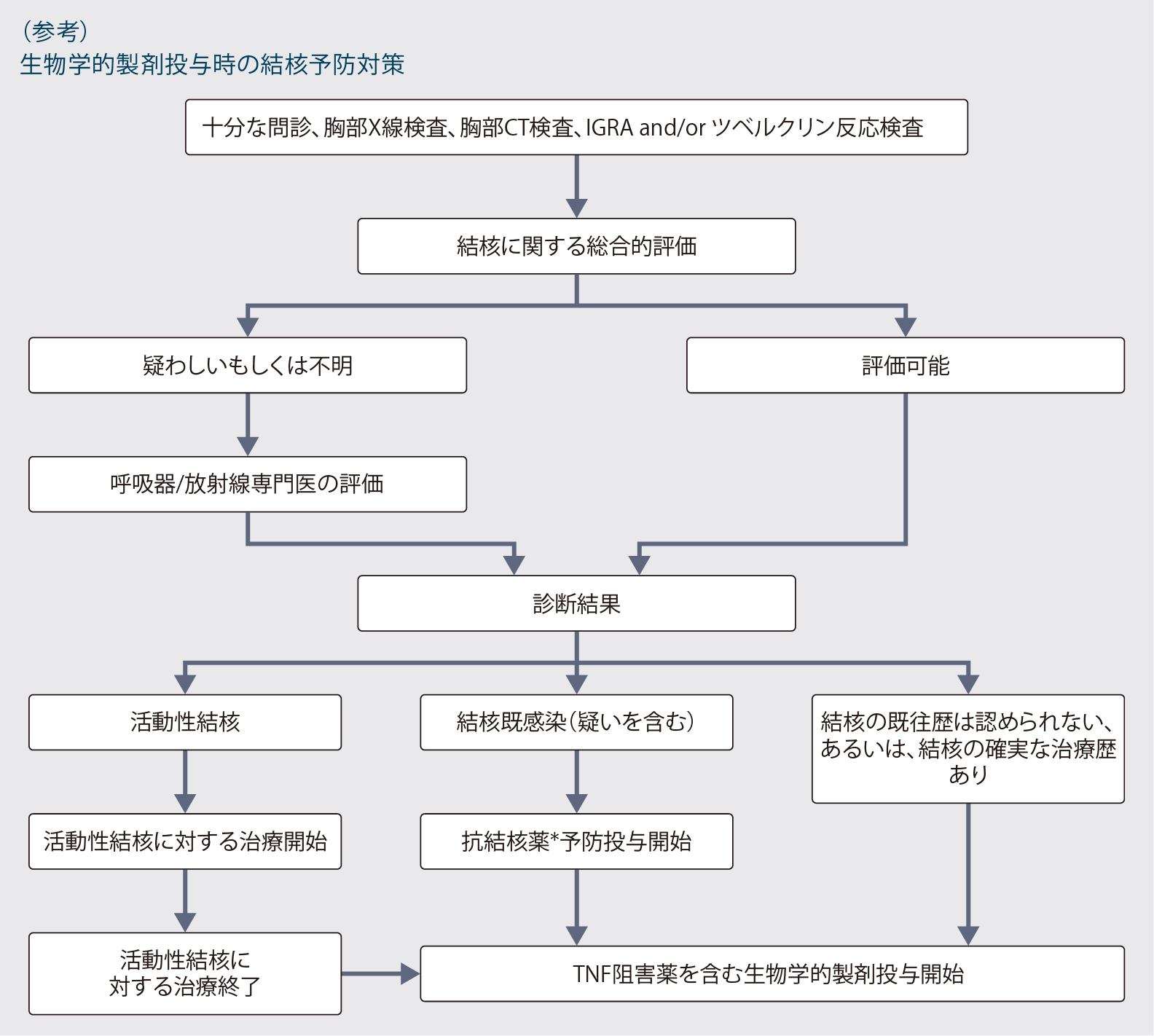
*:TNF阻害薬投与に先立つ3週間,抗結核薬(INHなど)の投与を行い,以後も計6~9カ月間並行して投与。
日本呼吸器学会 炎症性疾患に対する生物学的製剤と呼吸器疾患 診療の手引き 第2版作成委員会 編. 炎症性疾患に対する生物学的製剤と呼吸器疾患 診療の手引き 第2版, 2020
4)脱髄疾患(頻度不明※1)
- 本剤を含む抗TNF製剤投与により、脱髄疾患(多発性硬化症等)の臨床症状・画像診断上の新たな発現若しくは悪化がみられることがあります。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 脱髄疾患(多発性硬化症等)及びその既往歴のある患者さんに対しては、本剤は投与禁忌です。
- 脱髄疾患が疑われる徴候を有する患者さんでは神経学的評価や画像診断等の検査を行い、慎重に危険性と有益性を評価した上で本剤適用の妥当性を検討してください。また、脱髄疾患の家族歴のある患者さんにおいても、脱髄疾患発現のおそれがあるため、適宜画像診断等の検査を実施してください。
- 投与後は十分に観察を行ってください。
対処法
- 本剤投与後は十分に観察を行い、異常が認められた場合には本剤の投与を中止するなどの適切な処置を行ってください。
1. 警告(抜粋)
1.3
脱髄疾患(多発性硬化症等)の臨床症状・画像診断上の新たな発現若しくは悪化が、本剤を含む抗TNF製剤でみられたとの報告がある。脱髄疾患(多発性硬化症等)及びその既往歴のある患者には投与しないこととし、脱髄疾患を疑う患者に投与する場合には、適宜画像診断等の検査を実施するなど、十分な観察を行うこと。[9.1.3、11.1.4参照]
2. 禁忌(次の患者には投与しないこと)(抜粋)
2.4
脱髄疾患(多発性硬化症等)及びその既往歴のある患者[症状の再燃及び悪化のおそれがある。][9.1.3、11.1.4参照]
9.特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.3
脱髄疾患が疑われる徴候を有する患者及び家族歴のある患者
(1)脱髄疾患が疑われる徴候を有する患者については、各患者で神経学的評価や画像診断等の検査を行い、慎重に危険性と有益性を評価した上で本剤適用の妥当性を検討し、投与後は十分に観察を行うこと。脱髄疾患発現のおそれがある。
(2)脱髄疾患の家族歴のある患者は、適宜画像診断等の検査を実施し、十分注意すること。脱髄疾患発現のおそれがある。[1.1、1.3、2.4、11.1.4参照]
11. 副作用(抜粋)
11.1
重大な副作用
11.1.4
脱髄疾患(頻度不明) 中枢神経系又は末梢神経系の脱髄疾患(多発性硬化症、視神経炎、横断性脊髄炎、ギラン・バレー症候群等)があらわれることがある。[1.1、1.3、2.4、9.1.3参照]
5)重篤な血液障害(頻度不明※1)
- 本剤投与により、汎血球減少症、白血球減少、好中球減少、血小板減少等の重篤な血液障害が発現したとの報告があります。また、重篤な血液障害の既往を有する患者さんでは、症状が悪化するおそれがあります。
- 異常が認められた場合には本剤の投与を中止するなどの適切な処置を行ってください。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 投与後は十分に観察を行ってください。
対処法
- 異常が認められた場合には本剤の投与を中止するなどの適切な処置を行ってください。
6)うっ血性心不全(頻度不明※1)
- 本剤投与により、うっ血性心不全が発現又は悪化することがあります。
- 本剤はうっ血性心不全患者さんを対象とした臨床試験を実施していませんが、他の抗TNF製剤におけるうっ血性心不全を対象とした臨床試験では、心不全症状の悪化、死亡率の上昇が報告されており、本剤を含む抗TNF製剤によりうっ血性心不全の症状を悪化させるおそれがあります。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- うっ血性心不全の患者さんに対しては、本剤は投与禁忌です。
- 投与後は十分に観察を行ってください。
対処法
- 異常が認められた場合には本剤の投与を中止するなどの適切な処置を行ってください。
2. 禁忌(次の患者には投与しないこと)(抜粋)
2.5
うっ血性心不全の患者[11.1.6、15.1.2参照]
9.特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.4
重篤な血液疾患(汎血球減少症、白血球減少、好中球減少、血小板減少等)の患者又はその既往を有する患者 症状が悪化するおそれがある。[11.1.5参照]
11. 副作用(抜粋)
11.1
重大な副作用
11.1.5
重篤な血液障害(頻度不明) 汎血球減少症、白血球減少、好中球減少、血小板減少等の重篤な血液障害があらわれることがある。[9.1.4参照]
11.1.6
うっ血性心不全(頻度不明) うっ血性心不全の発現又は悪化があらわれることがある。[2.5参照]
7)重篤なアレルギー反応(頻度不明※1)
- 本剤投与により、アナフィラキシー様症状等の重篤なアレルギー反応があらわれることがあります。本剤の初回投与後に発現した患者さんも報告されています。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 本剤投与前に十分な問診を行い、アレルギー歴、家族歴等を確認してください。
- 投与後は十分に観察を行ってください。
対処法
- 異常が認められた場合には本剤の投与を中止し、速やかに適切な処置を行ってください。
8)ループス様症候群(頻度不明※1)
- 本剤投与により、ループス様症候群があらわれることがあります。
※1 先行バイオ医薬品(シンポニー®)の関節リウマチを対象とした国内臨床試験、潰瘍性大腸炎※2を対象とした国内臨床試験及び国際共同試験(日本人症例のみ)に基づく副作用発現頻度
※2 本剤は潰瘍性大腸炎に対して国内未承認である。
予防
- 投与後は十分に観察を行ってください。
対処法
- 異常が認められた場合には本剤の投与を中止し、速やかに適切な処置を行ってください。
2. 禁忌(次の患者には投与しないこと)(抜粋)
2.3
本剤の成分に対し過敏症の既往歴のある患者
11. 副作用(抜粋)
11.1
重大な副作用
11.1.7
重篤なアレルギー反応(頻度不明) アナフィラキシー様症状等の重篤なアレルギー反応があらわれることがある。本剤初回投与後に発現した症例もある。
11.1.8
ループス様症候群(頻度不明) [8.6参照]
(2)副作用一覧
〈海外第III相臨床試験(AVT05-GL-C01試験、海外データ)〉
1)16週目までの安全性
主な副作用
16週目までに副作用は、本剤群では19/251例(7.6%)、先行バイオ医薬品群では28/251例(11.2%)に認められた。主な副作用(1%以上に発現)は、本剤群ではアスパラギン酸アミノトランスフェラーゼ増加4例(1.6%)、アラニンアミノトランスフェラーゼ増加3例(1.2%)、先行バイオ医薬品群では注射部位反応6例(2.4%)、気管支炎、咽頭炎、上気道感染各3例(1.2%)であった。
16週目までに認められた副作用(いずれかの群で1%以上に発現)(安全性解析対象集団)
| 本剤群 (n=251) | 先行バイオ医薬品群 (n=251) | |
|---|---|---|
| n(%) | n(%) | |
| 副作用発現例 | 19(7.6) | 28(11.2) |
| 感染症および寄生虫症 | 7(2.8) | 15(6.0) |
| 気管支炎 | 0 | 3(1.2) |
| 咽頭炎 | 0 | 3(1.2) |
| 上気道感染 | 0 | 3(1.2) |
| 臨床検査 | 6(2.4) | 8(3.2) |
| アラニンアミノトランスフェラーゼ増加 | 3(1.2) | 2(0.8) |
| アスパラギン酸アミノトランスフェラーゼ増加 | 4(1.6) | 1(0.4) |
| 一般・全身障害および投与部位の状態 | 2(0.8) | 7(2.8) |
| 注射部位反応 | 1(0.4) | 6(2.4) |
MedDRA v27.1
重篤な有害事象
16週目までに重篤な有害事象は、本剤群では4例(1.6%)に認められ、それらの重症度は中等度1例(0.4%)、重度3例(1.2%)であった。重度の重篤な有害事象の内訳は甲状腺の良性新生物、感染性胸水、非感染性髄膜炎各1例(0.4%)、中等度の重篤な有害事象の内訳はネフローゼ症候群1例(0.4%)であった。先行バイオ医薬品群では2例(0.8%)に認められ、それらの重症度は重度1例(0.4%)、死亡1例(0.4%)であった。重度の重篤な有害事象の内訳は腎仙痛1例(0.4%)、死亡の内訳は遠隔転移を伴う新生物1例(0.4%)であった。
投与中止に至った有害事象
16週目までに投与中止に至った有害事象は、本剤群では4例(1.6%)、先行バイオ医薬品群では1例(0.4%)に認められた。投与中止に至った有害事象の内訳は、本剤群では甲状腺の良性新生物、感染性胸水、非感染性髄膜炎、ネフローゼ症候群各1例(0.4%)、先行バイオ医薬品群では遠隔転移を伴う新生物、上腹部痛各1例(0.4%)であった。
死亡に至った有害事象
16週目までに死亡に至った有害事象は、先行バイオ医薬品群で遠隔転移を伴う新生物が1例(0.4%)に認められた。本剤群では死亡に至った有害事象は認められなかった。
2)16週目から52週目までの安全性
主な副作用
16週目から52週目までに副作用は、本剤群では15/223例(6.7%)、先行バイオ医薬品群-本剤群では12/112例(10.7%)、先行バイオ医薬品群-継続群では18/113例(15.9%)に認められた。主な副作用(1%以上に発現)は、本剤群では鼻咽頭炎3例(1.3%)、先行バイオ医薬品-本剤群ではアラニンアミノトランスフェラーゼ増加、アスパラギン酸アミノトランスフェラーゼ増加各2例(1.8%)、先行バイオ医薬品-継続群では鼻咽頭炎3例(2.7%)、咽頭炎、上気道感染症、口腔ヘルペス、尿路感染、尿中白血球陽性各2例(1.8%)であった。
16週目から52週目までに認められた副作用(いずれかの群で1%以上に発現)(安全性解析対象集団)
| 本剤群 (n=223) | 先行バイオ医薬品-本剤群 (n=112) | 先行バイオ医薬品-継続群 (n=113) | |
|---|---|---|---|
| n(%) | n(%) | n(%) | |
| 副作用発現例 | 15(6.7) | 12(10.7) | 18(15.9) |
| 感染症および寄生虫症 | 9(4.0) | 6(5.4) | 12(10.6) |
| 鼻咽頭炎 | 3(1.3) | 1(0.9) | 3(2.7) |
| 咽頭炎 | 1(0.4) | 1(0.9) | 2(1.8) |
| 上気道感染症 | 1(0.4) | 1(0.9) | 2(1.8) |
| 口腔ヘルペス | 0 | 1(0.9) | 2(1.8) |
| 尿路感染 | 1(0.4) | 0 | 2(1.8) |
| 臨床検査 | 2(0.9) | 3(2.7) | 4(3.5) |
| アラニンアミノトランスフェラーゼ増加 | 1(0.4) | 2(1.8) | 0 |
| アスパラギン酸アミノトランスフェラーゼ増加 | 1(0.4) | 2(1.8) | 0 |
| 尿中白血球陽性 | 0 | 0 | 2(1.8) |
MedDRA v27.1
重篤な有害事象
16週目から52週目までに重篤な有害事象は、本剤群では6例(2.7%)、先行バイオ医薬品-本剤群では2例(1.8%)、先行バイオ医薬品-継続群では7例(6.2%)に認められた。重篤な有害事象の内訳は、本剤群では肺炎、関節リウマチ、乳癌、慢性冠症候群、脛骨骨折、関節リウマチ関連間質性肺疾患各1例(0.4%)、先行バイオ医薬品-本剤群では喉頭炎、胃炎各1例(0.9%)、先行バイオ医薬品-継続群では肺炎2例(1.8%)、鼠径ヘルニア、臍ヘルニア、関節リウマチ、背部痛、子宮内膜腺癌、低ナトリウム血症各1例(0.9%)であった。
投与中止に至った有害事象
16週目から52週目までに投与中止に至った有害事象は、本剤群では2例(0.9%)、先行バイオ医薬品-本剤群では2例(1.8%)、先行バイオ医薬品群-継続群で5例(4.4%)に認められた。投与中止に至った有害事象の内訳は、本剤群では乳癌、咳嗽各1例(0.4%)、先行バイオ医薬品-本剤群では結核、好酸球増加症各1例(0.9%)、先行バイオ医薬品-継続群では潜伏結核、術後創感染、背部痛、筋骨格障害、子宮内膜腺癌、胸水各1例(0.9%)であった。
死亡に至った有害事象
16週時目から52週目までに死亡に至った有害事象は認められなかった。
3)試験概要
| 目的 | 中等度から重度の関節リウマチ(RA)患者を対象に、メトトレキサート(MTX)併用時の本剤と先行バイオ医薬品(EU)※1の有効性、安全性及び免疫原性を比較する。 | ||||
|---|---|---|---|---|---|
| 試験デザイン | 多施設共同、無作為化、二重盲検、実薬対照、並行群間、2群間比較試験 | ||||
| 対象 | 18-75歳までの中等度から重度の関節リウマチ(RA)患者のうち、スクリーニング前の12週間以上MTXを投与しており、直近4週間に週12.5-25mg#以上の安定用量で、試験期間中も安定用量を継続する予定*2の患者502例 | ||||
| 試験方法 | 本試験の試験期間はステージ1(初回投与から16週目まで)及びステージ2(16週目から52週目まで)で構成される。対象を、ベースラインのDisease Activity Score(DAS)28-C反応性蛋白(CRP)(5.1以下及び5.1超)で層別し、本剤群又は先行バイオ医薬品群のいずれかに1:1の割合で無作為割り付けした。 本剤群:ステージ1では本剤50mgを初回投与し、その後は12週目まで4週ごとに皮下投与した。16週目にレスポンダー/ノンレスポンダー*3の状態を評価し、レスポンダーの場合にはステージ2に移行した。 ステージ2では本剤50mgを16週目から48週目まで4週ごとに皮下投与した。ノンレスポンダーの場合には治験薬投与を中止し、24週目まで追跡評価した。 先行バイオ医薬品群:ステージ1では先行バイオ医薬品(EU)50mgを初回投与し、その後は4週ごとに12週目まで皮下投与した。16週目にレスポンダー/ノンレスポンダー の状態を評価した。レスポンダーの場合には、ステージ2に移行して、1:1の割合で先行バイオ医薬品-本剤群(本剤50mgを16週目から48週目まで4週ごとに皮下投与)又は先行バイオ医薬品-継続群〔先行バイオ医薬品(EU)50mgを16週目から48週目まで4週ごとに皮下投与〕のいずれかに無作為割り付けした。ノンレスポンダーの場合には治験薬投与を中止し、24週目まで追跡評価した。 | ||||
| 主要評価項目 | 16週目におけるDAS28-CRPのベースラインからの変化量(検証的解析項目) | ||||
| 副次評価項目 |
| ||||
| 解析計画 | [有効性の解析]有効性の解析は、治験薬を1回以上投与したすべての患者を含む最大の解析対象集団(FAS)を対象とした。
[安全性の解析] 安全性の解析は治験薬を1回投与した患者を含む安全性解析対象集団を対象に、有害事象*6の発現率及び件数を要約した。 [免疫原性の解析] 免疫原性の解析は、安全性解析対象集団を対象に、ADA及びnAbの有無を投与群別、治験期間別に集計した。陽性抗体発現率も投与群別、治験期間別に集計した。 |
# 本邦におけるメトトレキサートの関節リウマチに対する用法及び用量は以下のとおりである。
- 6. 用法及び用量(抜粋)〈関節リウマチ〉通常、1週間単位の投与量をメトトレキサートとして6mgとし、1週間単位の投与量を1回又は2〜3回に分割して経口投与する。分割して投与する場合、初日から2日目にかけて12時間間隔で投与する。1回又は2回分割投与の場合は残りの6日間、3回分割投与の場合は残りの5日間は休薬する。これを1週間ごとに繰り返す。なお、患者の年齢、症状、忍容性及び本剤に対する反応等に応じて適宜増減するが、1週間単位の投与量として16mgを超えないようにする。
- *1 先行バイオ医薬品(EU)は欧州で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。
- *2 MTXのさらなる増量に不耐容である場合には、MTXを週10mg以上の投与も適格とした。
- *3 DAS28-CRPのベースラインからの減少量が0.6超かつDAS28-CRPが5.1以下をレスポンダー、DAS28-CRPのベースラインからの減少量が0.6以下又はDAS28-CRPが5.1超をノンレスポンダーと定義した。
- *4 SDAIは圧痛関節数(28関節評価)+腫脹関節数(28関節評価)+患者による疾患活動性評価(10cm VAS:0~10)+医師による疾患活動性評価(10cm VAS:0~10)+CRPの和として算出される。
- *5 CDAIは圧痛関節数(28関節評価)+腫脹関節数(28関節評価)+患者による疾患活動性評価(10cm VAS:0~10)+医師による疾患活動性評価(10cm VAS:0~10)の和として算出される。
- *6 有害事象はICH国際医薬用語集(MedDRA)v27.1を用いてコーディングした。
社内資料:海外第III相臨床試験(AVT05-GL-C01試験)[承認時評価資料]
【ゴリムマブBS皮下注50mgシリンジ「F」の効能又は効果、効能又は効果に関連する注意】
4.
効能又は効果 既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)
5.
効能又は効果に関連する注意 過去の治療において、少なくとも1剤の抗リウマチ薬(生物製剤を除く)等による適切な治療を行っても、 疾患に起因する明らかな症状が残る場合に投与すること。