ゴリムマブとは
開発の経緯
ゴリムマブは、遺伝子組換え完全ヒト免疫グロブリンG1κ(IgG1κ)モノクローナル抗体であり、ヒト腫瘍壊死因子α(TNFα)の可溶型及び膜結合型の両方と高い親和性があり、安定した複合体を形成し、TNFαの受容体への結合を阻害する。TNFαの過剰発現は、関節リウマチに特徴的な関節の炎症及び構造的損傷の重要な要因である。ゴリムマブはTNFαを阻害することにより、これらの疾患の炎症及びその他の症状を軽減する。
ゴリムマブBS皮下注50mgシリンジ「F」(以下、本剤)は「既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)」、「中等症から重症の潰瘍性大腸炎の改善及び維持療法(既存治療で効果不十分な場合に限る)」を適応とする先行バイオ医薬品シンポニー®皮下注50mgシリンジ(一般名:ゴリムマブ(遺伝子組換え))のバイオ後続品としてAlvotech社により開発された。本邦では富士製薬工業株式会社が、Alvotech社との独占的パートナーシップの合意に基づき、「バイオ後続品の品質・安全性・有効性確保のための指針」1)を踏まえ、品質特性に関する試験及び非臨床試験を実施し、本剤と先行バイオ医薬品(EU、US及びJP)※である欧州で承認されたゴリムマブ製剤との同等性を検討した。
また、健康成人を対象とした臨床薬理試験(AVT05-GL-P01試験)及び中等度から重度の関節リウマチ患者を対象とした海外第Ⅲ相臨床試験(AVT05-GL-C01試験)を実施し、本剤と先行バイオ医薬品(EU)との臨床的同等性を検討した。
富士製薬工業株式会社は、品質、非臨床、臨床のそれぞれの試験によって得られたエビデンスを評価した結果、本剤と先行バイオ医薬品(JP)の同等性・同質性が示されたことから、特許期間及び再審査期間が満了している「既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)」を本剤の効能又は効果として2024年10月に製造販売承認申請を行い、2025年9月に承認を取得した。
| ※ | 先行バイオ医薬品(EU)は、欧州で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。 先行バイオ医薬品(US)は、米国で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。 先行バイオ医薬品(JP)は、本邦で承認されているシンポニー®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。 |
|---|
【ゴリムマブBS皮下注50mgシリンジ「F」の効能又は効果、効能又は効果に関連する注意】
4. 効能又は効果 既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)
5. 効能又は効果に関連する注意 過去の治療において、少なくとも1剤の抗リウマチ薬(生物製剤を除く)等による適切な治療を行っても、疾患に起因する明らかな症状が残る場合に投与すること。
特性
製品の治療学的特性
1.
本剤はゴリムマブ(遺伝子組換え)のバイオ後続品(バイオシミラー)である。
2.
中等度から重度の関節リウマチ患者を対象とした海外第Ⅲ相臨床試験(AVT05-GL-C01試験)により、先行バイオ医薬品(EU)※1との有効性における同等性が検証されている(検証的解析結果)。
3.
重大な副作用として、敗血症性ショック・敗血症(頻度不明)・肺炎(0.9%)等の重篤な感染症、間質性肺炎(0.5%)、結核(頻度不明)、脱髄疾患(頻度不明)、重篤な血液障害(頻度不明)、うっ血性心不全(頻度不明)、重篤なアレルギー反応(頻度不明)、ループス様症候群(頻度不明)が報告されている※2。
なお、主な副作用として、鼻咽頭炎、上気道感染、注射部位反応(紅斑、硬結、そう痒感、蕁麻疹等)が報告されている。
詳細については電子添文の副作用及び臨床成績の安全性の結果を参照。
製品の製剤学的特性
1.
可溶型及び膜結合型のTNFαに結合する遺伝子組換えヒト免疫グロブリンG1κ(IgG1κ)モノクローナル抗体であるゴリムマブとして日本初のバイオ後続品である。
2.
穿刺防止安全装置付きのプレフィルドシリンジ製剤である。
※1 先行バイオ医薬品(EU)は、欧州で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。
※2 副作用発現頻度は先行バイオ医薬品(シンポニー®)の臨床試験成績に基づく。
【ゴリムマブBS皮下注50mgシリンジ「F」の効能又は効果、効能又は効果に関連する注意】
4. 効能又は効果 既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)
5. 効能又は効果に関連する注意 過去の治療において、少なくとも1剤の抗リウマチ薬(生物製剤を除く)等による適切な治療を行っても、疾患に起因する明らかな症状が残る場合に投与すること。
有効成分に関する理化学的知見
一般名:
ゴリムマブ(遺伝子組換え)[ゴリムマブ後続1](JAN)
分子式:
C6530H10068N1752O2026S44(タンパク質部分)
分子量:
約150,000
本 質:
ゴリムマブ[ゴリムマブ後続1](以下、ゴリムマブ後続1)は、遺伝子組換え抗腫瘍壊死因子α(TNF-α)モノクローナル抗体であり、ヒトⅠgG1に由来する。
ゴリムマブ後続1は、Sp2/0細胞により産生される。
ゴリムマブ後続1は、456個のアミノ酸残基からなるH鎖(γ1鎖)2本及び215個のアミノ酸残基からなるL鎖(κ鎖)2本で構成される糖タンパク質(分子量:約150,000)である。
薬物動態
血清中濃度
(1)単回投与
1)日本人及び外国人健康成人への単回投与時の薬物動態(AVT05-GL-P01試験)(日本人及び外国人データ)2)
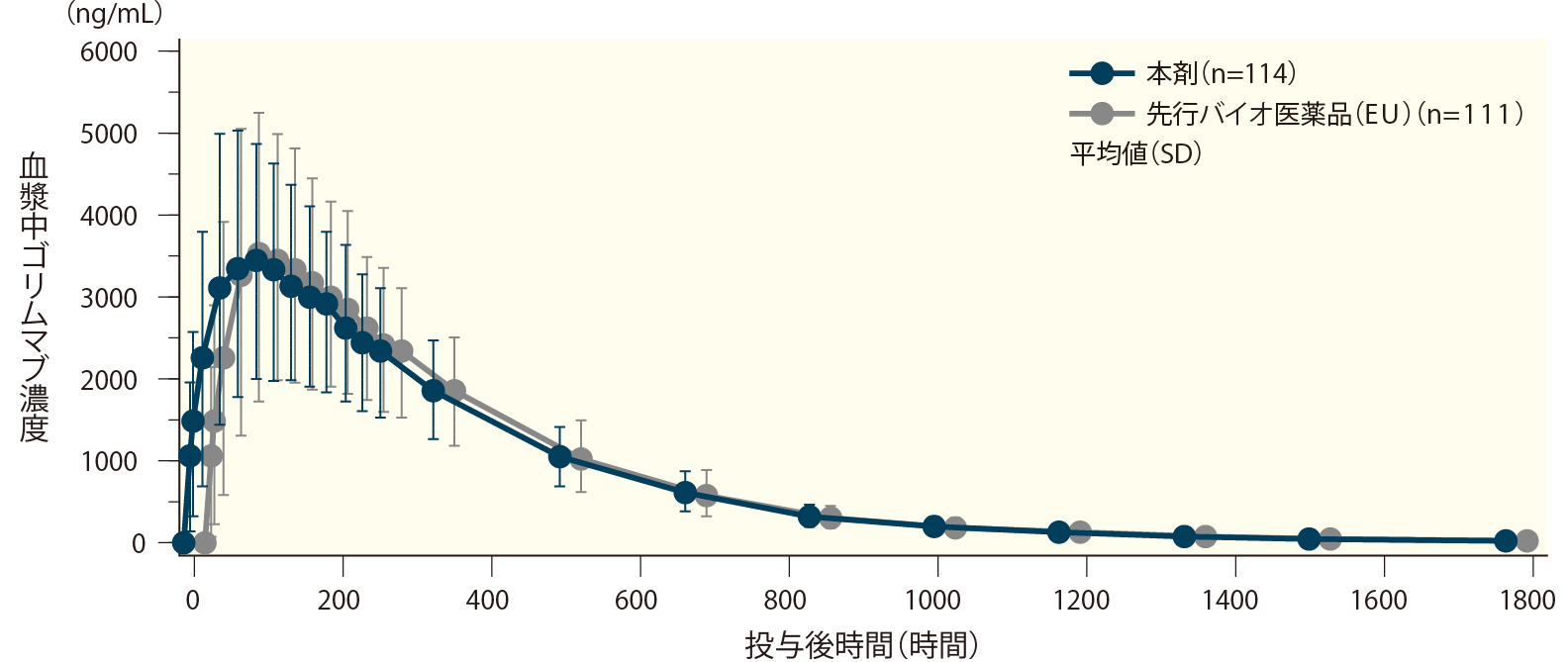
海外において、日本人を含む健康成人に本剤又は先行バイオ医薬品(EU)※をゴリムマブ(遺伝子組換え)として50mg単回皮下投与したときの血清中濃度の推移及び薬物動態(PK)パラメータを以下に示す。
PKパラメータのうち、0時間(投与前)から無限大に外挿した血清中濃度-時間曲線下面積(AUC0-inf)の幾何最小二乗(LS)平均値の比(90%信頼区間)は100.85%(92.33~110.15)、最高血清中濃度(Cmax)のLS平均値の比(90%信頼区間)は105.54%(98.46~113.13)、0時間(投与前)から時間tまでのAUC(AUC0-t)のLS平均値(90%信頼区間)は104.45%(97.36~112.06)で、いずれも生物学的同等性の基準範囲内(80~125%)であった。
※先行バイオ医薬品(EU)は欧州で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。
| 投与群 | AUC0-inf (時間·ng/mL) | AUC0-t (時間·ng/mL) | Cmax (ng/mL) | Tmax (時間) |
|---|---|---|---|---|
| 本剤 (n=114) | 1513406 ±504749.5 | 1484801 ±507013.4 | 3851.1 ±1697.34 | 96.03 (24-334.18) |
| 先行バイオ医薬品(EU) (n=111) | 1458286 ±516042.1 | 1439839 ±509836.1 | 3843.1 ±1661.02 | 95.75 (24-335.98) |
| 投与群 | Kel (1/日) | t1/2 (時間) | Vz/F (L) | CL/F (L/日) |
|---|---|---|---|---|
| 本剤 (n=114) | 0.0801 ±0.0277 | 232.78 ±80.242 | 12.3 ±5.754 | 0.91 ±0.38 |
| 先行バイオ医薬品(EU) (n=111) | 0.0774 ±0.02114 | 234.08 ±76.245 | 13.01 ±6.603 | 0.96 ±0.46 |
Tmax:最高血清中濃度到達時間、Kel:消失速度定数、t1/2:消失半減期、Vz/F:分布容積、CL/F:見かけの総血清クリアランス
平均値(SD)、Tmaxは中央値(範囲)
| 幾何最小二乗 平均値 | 幾何最小二乗 平均値の比(%) | 90%信頼区間 (%)*1,2 | ||
|---|---|---|---|---|
| Cmax(ng/mL) | ||||
| 本剤 (n=114) | 3578.11 | 100.85 | 92.33~110.15 | |
| 先行バイオ医薬品(EU) (n=111) | 3547.97 | |||
| AUC0-inf (時間·ng/mL) | ||||
| 本剤 (n=113) | 1455246.32 | 105.54 | 98.46~113.13 | |
| 先行バイオ医薬品(EU) (n=110) | 1378869.21 | |||
| AUC0-t (時間·ng/mL) | ||||
| 本剤 (n=114) | 1423690.74 | 104.45 | 97.36~112.06 | |
| 先行バイオ医薬品(EU) (n=111) | 1362997.20 | |||
*1 Cmax、AUC0-inf又はAUC0-tの自然対数変換値をもとに、投与群を固定効果、性別を因子、ベースラインの体重を連続共変量とした共分散分析モデルにより算出した。
*2 幾何最小二乗平均値の比の90%信頼区間が、事前に規定した範囲(80〜125%)の同等性マージン内に収まっている場合、同等であると判断した。
【ゴリムマブBS皮下注50mgシリンジ「F」の効能又は効果、効能又は効果に関連する注意】
4. 効能又は効果 既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)
5. 効能又は効果に関連する注意 過去の治療において、少なくとも1剤の抗リウマチ薬(生物製剤を除く)等による適切な治療を行っても、疾患に起因する明らかな症状が残る場合に投与すること。
6. 用法及び用量 〈メトトレキサートを併用する場合〉通常、成人にはゴリムマブ(遺伝子組換え)[ゴリムマブ後続1]として50mgを4週に1回、皮下注射する。なお、患者の状態に応じて1回100mgを使用することができる。〈メトトレキサートを併用しない場合〉通常、成人にはゴリムマブ(遺伝子組換え)[ゴリムマブ後続1]として100mgを4週に1回、皮下注射する。
2)健康成人男性への単回投与時の薬物動態
健康成人男性に本剤50mg及び100mgを単回皮下投与したときの血清中ゴリムマブ濃度は投与後3.50~5.50日後に最高濃度に達し、約12~13日の消失半減期で低下した。血清中ゴリムマブのCmax及びAUC∞は、50mg及び100mgの用量範囲において用量にほぼ比例して増加した3-6)。
| 用量 | 50mg (N=12) | 100mg (N=12) |
|---|---|---|
| Cmax(μg/mL) | 2.82±0.97 | 6.72±2.35 |
| Tmax(day) | 5.50(3.00, 10.07) | 3.50(2.00, 7.01) |
| AUC∞(μg・day/mL) | 53.25±13.06 | 121.63±33.89 |
| CL/F(mL/day/kg) | 15.21±3.88 | 13.41±3.74 |
| Vdz/F(mL/kg) | 256.73±60.94 | 237.00±57.98 |
| t1/2(day) | 11.92±2.32 | 12.56±2.41 |
Tmax:中央値(最小値、最大値)
健康成人男性に本剤200mgを単回皮下投与したときの血清中ゴリムマブ濃度は投与後5.00日に最高濃度に達し、約12日の消失半減期で低下した6)。
| (N=11) | |
|---|---|
| Cmax(μg/mL) | 15.9±3.0 |
| Tmax(day) | 5.0(3.0, 6.0) |
| AUC∞(μg・day/mL) | 268.9±51.8 |
| t1/2(day) | 12.3±1.5 |
Tmax:中央値(最小値、最大値)
(2)反復投与
日本人関節リウマチ患者に4週ごとに本剤50mg又は100mgを反復皮下投与したとき、血清中ゴリムマブ濃度は投与開始12週目までに定常状態に達した。血清中ゴリムマブのトラフ濃度は用量にほぼ比例して増加した7)。
(3)吸収(外国人データ)
健康成人男性に本剤100mgを単回皮下投与したときの絶対的バイオアベイラビリティは51%であった。上腕部、腹部及び大腿部に皮下投与したとき、絶対的バイオアベイラビリティは投与部位間で差はなかった7)(外国人データ)。
(4)代謝
ゴリムマブは、ヒトⅠgG1モノクローナル抗体であることから、他の免疫グロブリンG1と同様8)に代謝されると推察される9)。
薬効薬理
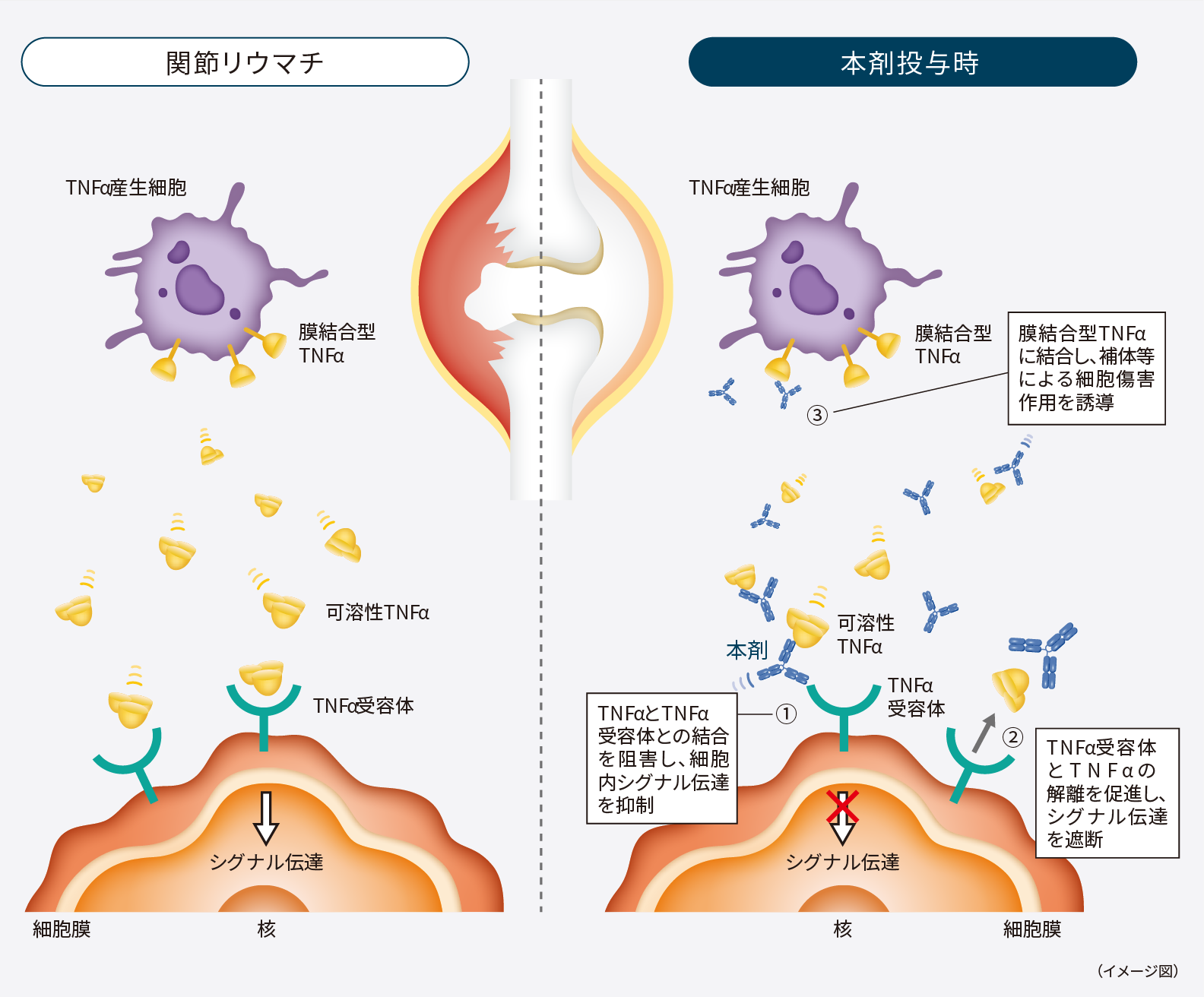
1.作用機序
本剤はin vitro試験において、可溶性及び膜結合型TNFαに対して選択的に結合し、以下の作用を示した10,11)。
- TNFαのTNF受容体への結合を阻害した。
- TNFα刺激による線維芽細胞又は内皮細胞のサイトカイン(IL-6、IL-8、G-CSF、GM-CSF)の産生及び内皮細胞での接着分子(E-セレクチン、ICAM-1、VCAM-1)の発現を抑制した。
ゴリムマブ「F」の作用機序
2.非臨床試験
(1)可溶性及び膜結合型TNFα中和作用(in vitro)12)
本剤及び先行バイオ医薬品(EU、US及びJP)の中和作用を以下に示す。
| 相対TNFα中和作用 (標準品に対する活性比) | |
|---|---|
| 本剤 (n=4) | 97~100% |
| 先行バイオ医薬品 (EU)(n=9) | 91~106% |
| 先行バイオ医薬品 (US)(n=6) | 88~104% |
| 先行バイオ医薬品 (JP)(n=3) | 90~98% |
(2)可溶性及び膜結合型TNFα結合作用(in vitro)12)
本剤及び先行バイオ医薬品(EU、US及びJP)の可溶性及び膜結合型TNFα結合作用を以下に示す。
| 可溶性TNFα相対結合親和性 (標準品に対する活性比) | 膜結合型TNFα相対結合親和性 (標準品に対する活性比) | |
|---|---|---|
| 本剤 (n=4) | 97.0~105.4% | 95.0~103.0% |
| 先行バイオ医薬品 (EU)(n=9) | 82.5~107.4% | 79.2~121.7% |
| 先行バイオ医薬品 (US)(n=6) | 88.8~116.4% | 89.5~105.5% |
| 先行バイオ医薬品 (JP)(n=3) | 95.8~97.1% | 103.0~108.0% |
(3)ADCC活性(in vitro)13)
本剤及び先行バイオ医薬品(EU、US及びJP)のADCC活性を以下に示す。
| 相対ADCC活性 (標準品に対する活性比) | |
|---|---|
| 本剤 (n=4) | 104~115% |
| 先行バイオ医薬品 (EU)(n=9) | 95~138% |
| 先行バイオ医薬品 (US)(n=6) | 97~142% |
| 先行バイオ医薬品 (JP)(n=3) | 125~130% |
(4)CDC活性(in vitro)13)
本剤及び先行バイオ医薬品(EU及びUS)のCDC活性を以下に示す。
| 相対CDC活性 (標準品に対する活性比) | |
|---|---|
| 本剤 (n=4) | 91~97% |
| 先行バイオ医薬品 (EU)(n=9) | 94~122% |
| 先行バイオ医薬品 (US)(n=6) | 85~119% |
※先行バイオ医薬品(EU)は、欧州で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。
先行バイオ医薬品(US)は、米国で承認されているSimponi®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。
先行バイオ医薬品(JP)は、本邦で承認されているシンポニー®〔ゴリムマブ(遺伝子組換え)製剤〕を指す。
3.抗リウマチ作用(マウス)
本剤は、ヒトTNFαトランスジェニックマウスの関節炎の発症を有意に遅延し、関節の病理組織学的変化を有意に抑制した14)。
安全性薬理試験及び毒性試験
日本、欧州及び米国のバイオ後続品の開発に関して適用される規制ガイドラインに従い、安全性薬理試験、毒性試験は実施していない。
- バイオ後続品の品質・安全性・有効性確保のための指針」(厚生労働省)(https://www.pmda.go.jp/files/000266866.pdf)(2025年10月参照)
- 社内資料:海外第Ⅰ相臨床試験成績(AVT05-GL-P01)
- Ling J, et al.:J Clin Pharmacol. 2010; 50: 792-802
- 個々の試験結果の要約(シンポニー皮下注50mgシリンジ: 2011年7月1日承認、申請資料概要2.7.2.2)
- 全試験を通しての結果の比較と解析(シンポニー皮下注50mgシリンジ:2011年7月1日承認、申請資料概要2.7.2.3)
- 日本人及び白人健康成人を対象とした海外第Ⅰ相試験(シンポニー皮下注50mgシリンジ:2011年7月1日承認、審査報告書)
- Xu Z, et al.:J Clin Pharmacol. 2010; 50: 276-284
- Tabrizi MA, et al.:Drug Discov Today. 2006; 11: 81-88
- 代謝(シンポニー皮下注50mgシリンジ:2011年7月1日承認、申請資料概要2.4.3.4)
- ヒトTNFαに対する結合能の解析(シンポニー皮下注50mgシリンジ:2011年7月1日承認、申請資料概要2.6.2.1.1.1)
- ヒトTNFα活性の抑制機序(シンポニー皮下注50mgシリンジ:2011年7月1日承認、申請資料概要2.6.2.1.1.2)
- 社内資料:薬理作用に関連する試験―可溶性及び膜結合型TNFαに対する結合活性―
- 社内資料:薬理作用に関連する試験―ADCC及びCDC活性―
- マウス多発性関節炎モデルに対する作用(シンポニー皮下注50mgシリンジ:2011年7月1日承認、申請資料概要2.6.2.1.1.5)